Alport综合征:隐藏在基因里的“肾脏杀手”
作者:张琰琴 北京大学第一医院 副研究员
审核:丁洁 北京大学第一医院 教授
Alport综合征是一种遗传性肾脏疾病,以首先报道该病的医生姓氏命名。Alport综合征(也叫遗传性进行性肾炎)虽属罕见病,却需要高度警惕,因为它会悄无声息地导致肾功能衰竭。
随着诊断水平不断提高,临床确诊的病例逐渐增多。不是发病率增加了,而是诊断能力提升的结果。同时,了解Alport综合征的遗传方式对家庭生育指导至关重要,通过遗传咨询和产前诊断,可有效避免患儿出生。
Alport综合征有四种遗传方式,遗传规律各不相同。
X连锁遗传(性连锁遗传)最常见,约占80%-85%,致病基因位于X染色体上。男性患者症状重,因只有一条X染色体,基因突变后无正常基因替代;女性患者症状轻,因有两条X染色体,一条异常一条正常。女性患者的子女有50%概率遗传(50%儿子患病,症状重;50%女儿患病,症状轻)。男性患者传女不传男(女儿100%为患者,症状轻;儿子不遗传)。
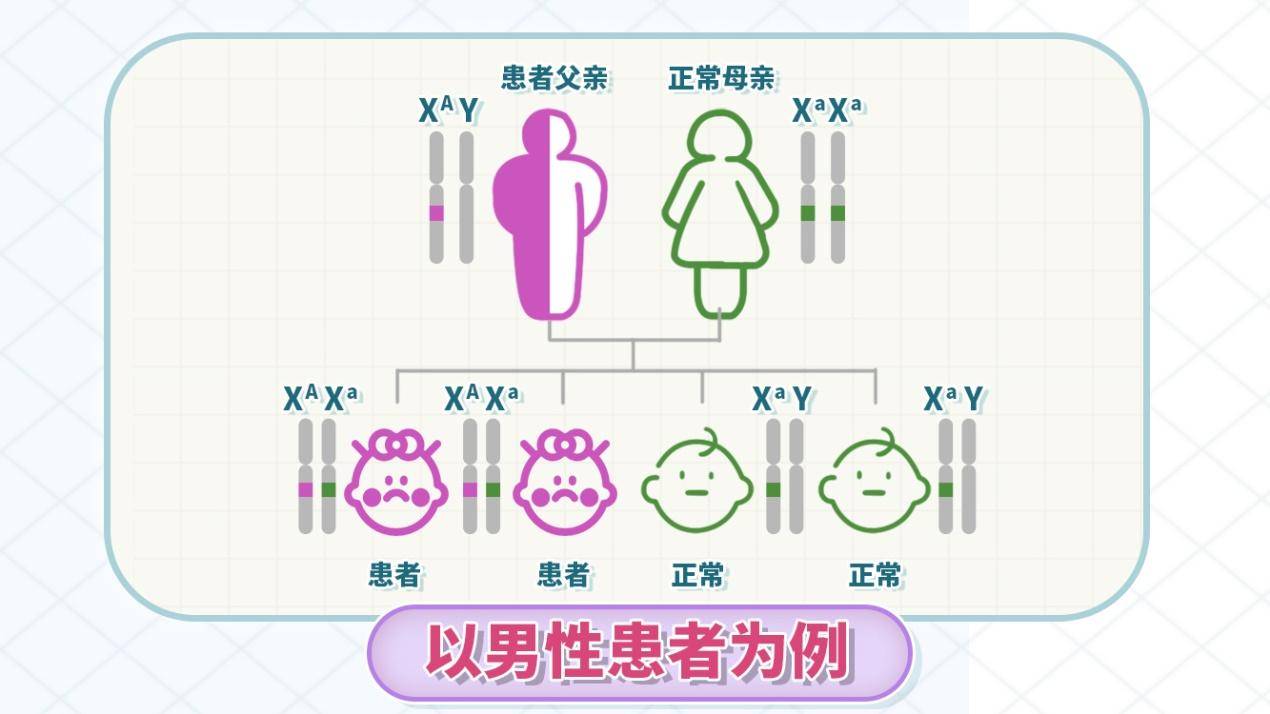
图1 原创版权图片,不授权转载
常染色体隐性遗传男女患者的患病率和症状无差异,父母通常为无症状携带者,子女患病概率为25%。
常染色体显性遗传每代均有患者,子女患病概率为50%。
双基因遗传占比最少,约占1%,遗传规律需根据突变基因具体分析。
对于有家族史的人群,建议进行遗传咨询和基因检测,明确携带状态,科学规划生育。
Alport综合征其本质是IV型胶原蛋白基因突变导致肾脏滤过膜结构异常。肾脏如同身体的排污工厂,肾小球基底膜则是关键的过滤筛网,IV型胶原是构成这个筛网的核心材料。基因突变导致IV型胶原蛋白合成异常,就像用次品线绳织网,初期筛网虽有缺陷尚能工作,但随着时间推移,筛网逐渐破损,最终完全失去过滤功能,导致尿毒症。具体而言,COL4A5基因突变导致X连锁遗传;COL4A3或COL4A4基因突变导致常染色体遗传。
Alport综合征的症状通常在儿童期开始显现,呈渐进性发展。血尿通常是最早出现的症状,多为镜下血尿(肉眼不可见,需化验发现)。部分患儿可出现肉眼血尿,尿液呈浓茶色。随病情进展出现蛋白尿,提示肾损伤加重,最终发展为尿毒症,通常在青壮年时期就可能需要肾脏替代治疗。
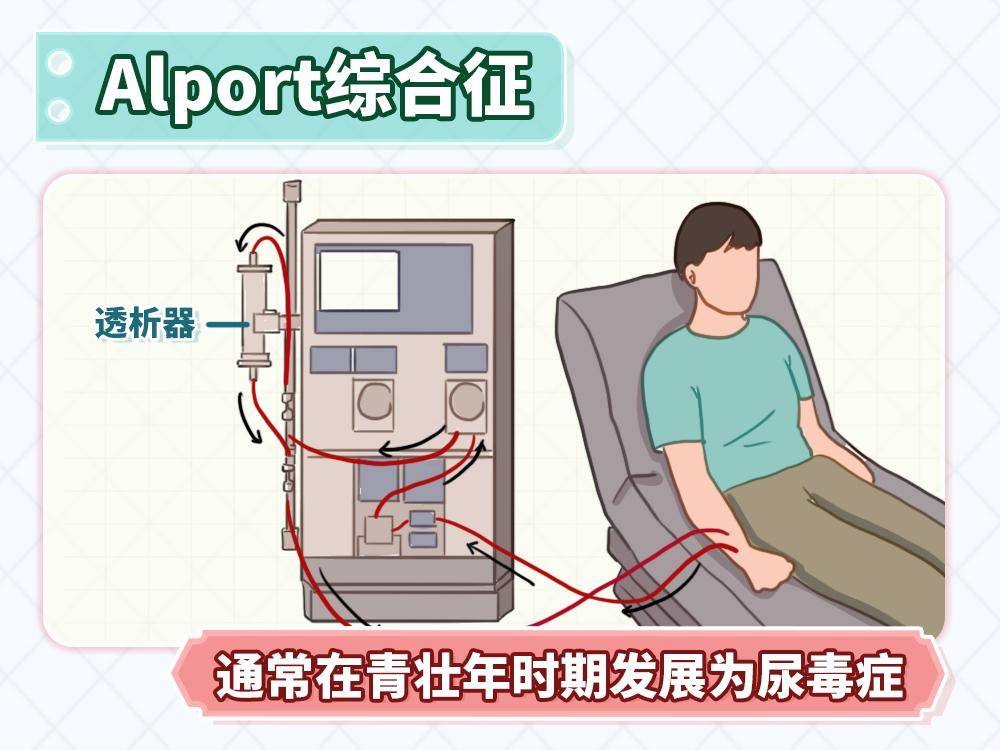
图2 原创版权图片,不授权转载
不同遗传方式预后差异显著,尤其X连锁遗传存在明显性别差异。X连锁遗传患者男性几乎100%进展至尿毒症,多在20-30岁需要肾脏替代治疗。女性多数症状较轻,约15%-20% 会发展为尿毒症,且发病年龄较晚(40-60岁)。虽然女性患者总体预后较好,但绝不能掉以轻心,仍需定期随访,及时发现并干预危险因素。常染色体遗传患者男女预后相似,取决于具体基因突变类型。
Alport综合征除了肾脏表现外,还有肾外表现,可作为辅助诊断的线索。需要注意的是,并非所有患者都有肾外表现,但肾脏受累是必然的。约半数患者出现感音神经性耳聋,早期需专业听力检测才能发现;眼睛可以有前圆锥形晶状体、视网膜病变等,表现为进行性近视且难以矫正;食管、气管等部位出现弥漫性平滑肌瘤,引起吞咽困难等症状。耳聋和眼部病变的发生率约50%-60%,平滑肌瘤则非常罕见。
Alport综合征的早期诊断依赖于对疑似症状的警惕,如儿童期持续不明原因血尿;家族中有肾脏病或耳聋病史;进行性听力下降或特殊眼部病变。早期诊断、早期干预可显著延缓尿毒症发生,为患者争取更长时间的正常生活。随着医疗技术进步,即使进入尿毒症期,通过透析或肾移植,患者仍可长期生存,同样可以拥有有意义的人生。

图文简介